GtoPdb is requesting financial support from commercial users. Please see our sustainability page for more information.
|
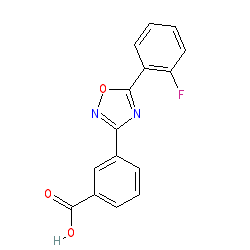
Synonyms: PTC124

ataluren is an approved drug (EMA (2014))
Compound class:
Synthetic organic
Comment: Ataluren is a compound in an emerging class of chemicals which induce ribosomal readthrough. This developing pharmaceutical intervention has been termed 'readthrough strategy'. Clinically, this type of compound has potential benefit in the treatment of genetic disorders associated with loss of functional protein due to nonsense or PTC mutations, such as certain types of muscular dystrophy, cystic fibrosis and many cancers [1,4].
|
|
|||||||||||||||||||||||||||||||||||
| No information available. |
Summary of Clinical Use  |
| This drug was granted orphan designation by the European Medicines Agency (EMA) in 2005, and full apporval in 2014 for the treatment of Duchenne muscular dystrophy (DMD). Many clinical trials have been further assessing the efficacy of this drug in patients with nonsense mutation dystrophinopathies. Results from clinical trials testing ataluren have been reported [2] and more trials are underway. Click here to view current clinical trials of ataluren/PTC124 registered at ClinicalTrials.gov. Trial NCT02090959 is a confirmatory Phase 3 extension study in 220 patients with nonsense mutation dystrophinopathy which is a 96 week trial due for completion in mid-June 2014. Ataluren is also being investigated for use in patients with nonsense mutation cystic fibrosis in Phase 3 studies (NCT00803205 and NCT01140451). A Phase 2a study in patients with nonsense mutation hemophilia A and B (NCT00947193) has been suspended. A note of caution: The US FDA has rejected applications for approval of ataluren as it has repeatedly failed to show statistically significant results, failing both a Phase 2b study for DMD as well as a Phase 3 study for cystic fibrosis. The number of failures in late stage trials suggests that the only conclusion to be made is that ataluren is not clinically effective, despite being based on a sound biological mechanism. Read the In The Pipeline blogpost 'Bad News-But Not the Unexpected Kind' for further comments and debate around ataluren's future. |
| Mechanism Of Action and Pharmacodynamic Effects |
| Studies have shown that the mechanism of action involves suppression of ribosome-induced termination of protein translation at premature termination codons (PTCs) whilst still recognising genuine stop codons [4]. By increasing the ability of the ribosome to ignore PTCs, increased amounts of functional full-length proteins are produced. However, the validity of the claimed action of this chemical has been questioned [3]. In Duchenne muscular dystrophy (DMD) the dystrophin gene carries a PTC in around 10 to 15% of patients. |
| Clinical Trials | |||||
| Clinical Trial ID | Title | Type | Source | Comment | References |
| NCT00803205 | Study of Ataluren (PTC124™) in Cystic Fibrosis | Phase 3 Interventional | PTC Therapeutics | ||
| NCT00947193 | Study of Ataluren (PTC124®) in Hemophilia A and B | Phase 2 Interventional | PTC Therapeutics | ||
| NCT01140451 | Extension Study of Ataluren (PTC124) in Cystic Fibrosis | Phase 3 Interventional | PTC Therapeutics | ||
| NCT02090959 | An Extension Study of Ataluren (PTC124) in Participants With Nonsense Mutation Dystrophinopathy | Phase 3 Interventional | PTC Therapeutics | ||