GtoPdb is requesting financial support from commercial users. Please see our sustainability page for more information.
|
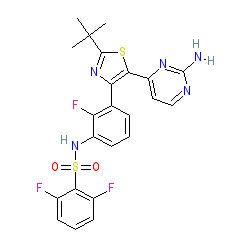
Synonyms: GSK2118436 | GSK2118436A | Tafinlar®

dabrafenib is an approved drug (FDA and EMA (2013))
Compound class:
Synthetic organic
Comment: Dabrafenib is a Type-1.5 kinase inhibitor.
Ligand Activity Visualisation ChartsThese are box plot that provide a unique visualisation, summarising all the activity data for a ligand taken from ChEMBL and GtoPdb across multiple targets and species. Click on a plot to see the median, interquartile range, low and high data points. A value of zero indicates that no data are available. A separate chart is created for each target, and where possible the algorithm tries to merge ChEMBL and GtoPdb targets by matching them on name and UniProt accession, for each available species. However, please note that inconsistency in naming of targets may lead to data for the same target being reported across multiple charts. ✖
View more information in the IUPHAR Pharmacology Education Project: dabrafenib |
|
|||||||||||||||||||||||||||||||||||
| No information available. |
Summary of Clinical Use  |
| Dabrafenib is a kinase inhibitor approved for the treatment of patients with unresectable or metastatic melanoma with BRAF mutations as detected by an FDA-approved test and should not be used to treat patients with wildytpe BRAF tumors. Marketed formulations contain dabrafenib mesylate (PubChem CID 44516822). In April 2018, the FDA approved the use of dabrafenib in combination with trametinib for the adjuvant treatment of patients with BRAF V600E or V600K mutated melanoma (as detected by an FDA-approved test) and involvement of lymph node(s), following complete resection. Then in May, this same combination was approved (FDA) for the treatment of locally advanced/metastatic BRAF V600E +ve anaplastic thyroid cancer. The EMA issued orphan drug designation in November 2023, for the treatment of low- and high-grade gliomas with a BRAF V600E mutation in combination with trametinib. |
| Mechanism Of Action and Pharmacodynamic Effects |
| Dabrafenib potently inhibits (IC50 in low nanomolar concentrations) constitutively active BRAF kinase with mutations V600E (VAR_018629)., V600K, and V600D in vitro. The mechanism of tumor inhibition is thought to be via dabrafenib-induced inhibition of phosphorylated ERK resulting in a decrease in cell proliferation. Downstream mediators of the MAPK pathway are also inhibited. Dabrafenib also inhibits wild-type BRAF and CRAF kinases at low IC50 concentrations and other kinases such as SIK1, NEK11, and LIMK1 at higher concentrations. |
| Pharmacokinetics |
| Absorption/Distribution |
| Peak plasma concentration is reached 2 hours after oral administration. It is recommended to dose in the fasted state. Mean absolute bioavailability of oral dabrafenib is 95%. Dabrafenib is 99.7% bound to human plasma proteins. |
| Biotransformation/Metabolism |
| Dabrafenib is excreted in urine and bile as carboxy-dabrafenib (by the sequential actions of CYP2C8 and CYP3A4). Desmethyl-dabrafenib is a decarboxylation metabolite that may be reabsorbed from the gut. The parent drug and the hydroxy- metabolite have similar terminal half-lives (10 hours), but the carboxy- and desmethyl- metabolites have extended half-lives (21-22 hours). Both hydroxy- and desmethyl-dabrafenib are likely to contribute to the clinical activity of dabrafenib. |
| Elimination |
| Excretion of dabrafenib is primarily via feces (71%) with urinary excretion accounting for 23% ( recovered as metabolites only). |
| Population pharmacokinetics |
| There are no clinically relevant differences in exposure to dabrafenib associated with age, gender or weight. The pharmacokinetics of dabrafenib have not been studied in pediatric patients. |
| Organ function impairment |
| Mild or moderate renal impairment has no effect on systemic exposure to dabrafenib and its metabolites. No data are available in patients with severe renal impairment. Mild hepatic impairment has no effect on systemic exposure to dabrafenib and its metabolites. No data are available in patients with moderate to severe hepatic impairment. |
| External links |
|
For extended ADME data see the following: Electronic Medicines Compendium (eMC) Drugs.com European Medicines Agency (EMA) |